Merohedral twin
refinement in GSAS-II
Introduction:
In these exercises you will use GSAS-II to refine the
structure of a few single crystal structures where there is merohedral
twinning. There is a general discussion of twinning in International Tables for
Crystallography (2006), Vol. C, Chapter 1.3, pp 10-14. Merohedral
twinning occurs when the twin operation (2, m or ![]() )
belongs to the point group of the vector lattice but not of the crystal
structure. Of the 230 space groups only 159 of them may have merohedral
twining.
)
belongs to the point group of the vector lattice but not of the crystal
structure. Of the 230 space groups only 159 of them may have merohedral
twining.

The possible merohedral twin operators are shown in the above table (extracted from p 13 of Intl. Tables for Cryst., Vol. C). Merohedral twins are of two types: Type 1 are inversion twins and have only 2 components and Type 2 are where the twin operation does not belong to the Laue class of the crystal (only for space groups marked with * in above table) and multiple twin components may be found. For Type 1 the twin lattice exactly superimpose so that if Friedel’s law is obeyed the twin components have the same value of |F|2 (the twinning is undetectable!). However, in the presence of resonant scattering the |F|2 components differ and if the twin components have unequal volumes then the Bragg intensities will differ from that obtained from the untwinned structure and then the twinning effects must be considered for refinement. Obviously all Type 1 crystals are noncentrosymmetric. For Type 2 the space group may be centrosymmetric from within a choice of just 55 possible space groups. However, the diffraction pattern may display apparent higher symmetry especially if the twin components are all equally present in the crystal. Note that menu entries and user input are shown in bold face below as Help/About GSAS-II, which lists first the name of the menu (here Help) and second the name of the entry in the menu (here About GSAS-II). If you have not done so already, start GSAS-II.
.
Example 1. A Type 1
inversion twin – C8H25Al0.84Ga1.16N3
This example was taken from Luo, et al., Dalton Trans. 4491(2006). The structure is given in the file MerohedralTwins/data/1b.cif and the data are in MerohedralTwins/data/1b.fcf. This example is noncentrosymmetric and the data was collected with radiation (MoKα) that gives significant resonant scattering from the Ga atoms.
Step 1. Setup for
refinement
To begin do Import/Phase/from CIF file from the main GSAS-II data tree menu. A file selection window will appear; find the above cif file (…1b.cif) and select it. A popup window will appear. Press Yes; a new popup will appear; Press OK, and you will see the General page for this phase
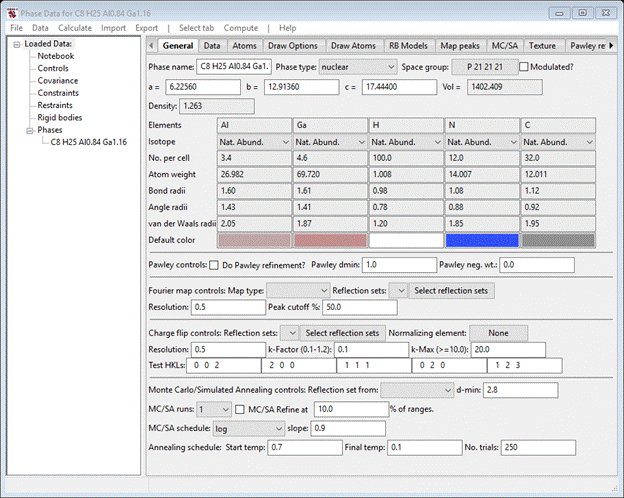
Next do Import/Structure Factor/from CIF file (that is the format of a “fcf” file); a file selection dialog box will appear with the previously selected directory chosen. Select 1b.fcf; a popup window will appear
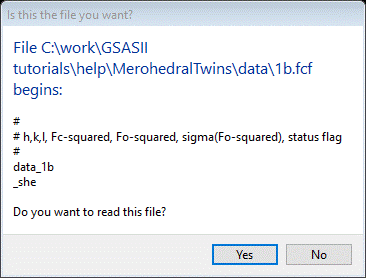
Select Yes; a new popup window allowing you to change the name
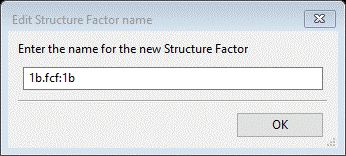
Press OK; after a pause and some activity a popup window will appear

This makes the connection between the data set and the previously imported phase. Select the entry and press OK. You now have loaded all that is needed for the first example. The structure had previously been solved and refined with Shelx so you need not trouble about those steps. You can try to solve the structure in GSAS-II via charge flipping but this is not part of this exercise; we will only explore the twin analysis features of GSAS-II.
Step 2. Refinement
of twinned structure
Before beginning the refinement, it usually the practice to eliminate the poorly measured reflections where F2obs < 2s(F2). Select the Controls entry from the GSAS-II data tree.
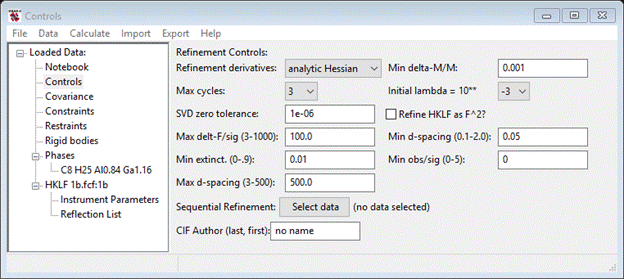
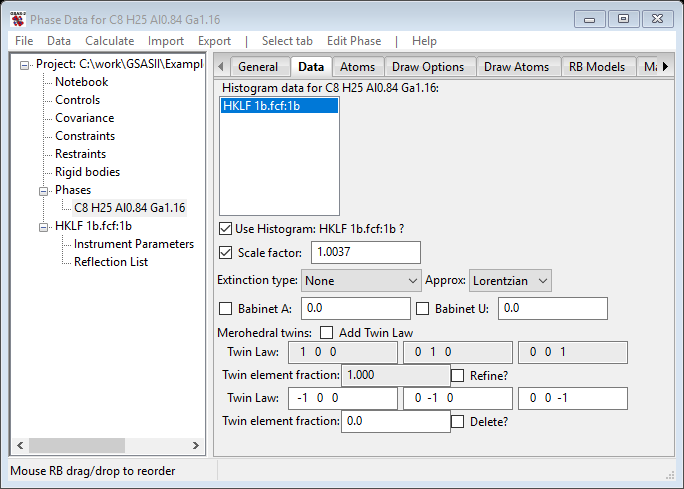
Change Min obs/sig to 2.0. Next, select the Phases/C8 H25 Al0.84 Ga1.16 entry in the GSAS-II data tree and choose the Data tab
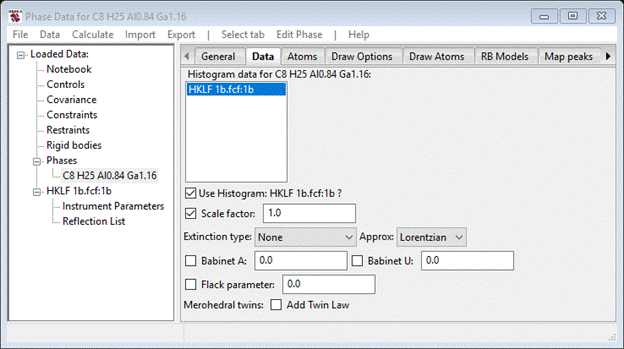
Notice that the Scale factor is selected for refinement by default. If you refine it, you will get an Rw= 2.85% which isn’t too bad. However, you are unsure what the hand of the molecule is in this structure and the experiment with MoKa radiation should give sufficient resonant scattering to determine this. The Flack parameter gives you a way to evaluate which hand the molecule is. If upon refinement the value is ~0 then you have correctly described the hand. Otherwise if the value is ~1 then the opposite hand is the correct and the structure needs to be inverted. So let’s test this; select the Flack parameter for refinement and do Calculate/Refine from the main GSAS-II data tree menu. You’ll be asked to save the project; pick a directory & a name – I chose 1b. The structure will quickly refine to give Rw=2.155% which is a considerable improvement on Scale factor alone. The Flack parameter is now 0.433, meaning that both hands somehow exist in the structure. This can happen if the crystal is merohedrally inversion twinned.
To introduce this twin, press the Add Twin Law box; the window will be redrawn.

Notice that two Twin Law matrices have been generated; one is the identity for the reference structure and the other by default is the merohedral inverse. If some other twin law is desired, then the matrix elements need to be changed. Moreover, the Flack parameter has vanished as it is no longer needed. Press the Refine button for the Twin Law fraction and rerun the refinement. The Data window now has.
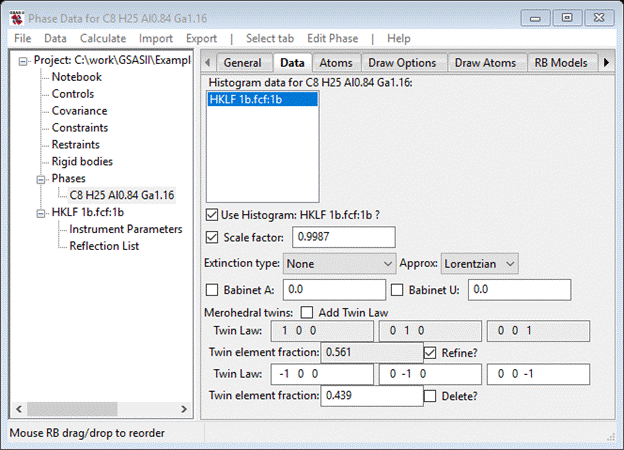
The refinement quickly converged to where it was before but the twin fractions are now ~56/44 consistent with the ~45% Flack parameter obtained earlier. It is likely that the crystal is macroscopically twinned thus the twin fractions are not exactly 50/50. This completes this example; you can save your project if you want to explore other features of GSAS-II with it.
Example 2. A Type 2
only merohedral twin –
[Al(OC6H4(C3H7)2)]-
[NC7H8]+
In this case the data collection Laue statistics (very nearly -3m) and systematic extinctions indicated that the space group was either one of the enantiomorphic pair P3121 or P3221 or possibly one of the pair P31 or P32. If the true structure was of the lower symmetry P31 or P32 then there must be Type 2 merohedral twinning to make the diffraction pattern appear to have higher symmetry (-3m instead of -3); this exercise will explore this possibility. The data were collected with MoKa radiation so there is little resonant scattering and thus the choice of hand is indeterminate. Solving the structure in P3221 yielded a poorly fitting structure with disorder particularly in the cation. Resolving the structure in P32 gave a structure with no disorder and a better fit to the data (but still not great). As this work was done with Shelx this result is in the form of a Shelx input file (2b.ins – thanks to V. Young for this example). If you have not done so already, start GSAS-II and start with a fresh project.
Step 1. Setup for
refinement
To begin we need to import the phase information from the Shelx ins file. From the main GSAS-II data tree menu, do Import/Phase/from Shelx ins, res file (the ins file is input to Shelx and a res file is the output result after a Shelx run; either can be used here). Select 2b.ins from the file selection window; a popup window will ask if this file is the one you want.
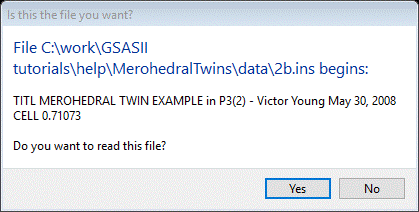
Press Yes; a Read Warning will appear.

There are two significant warnings concerning Shelx files. One is that the file never contains the space group symbol (Shelx requires just the operators), so GSAS-II sets the space group to P 1 by default and you will need to change it to P32 immediately (next window). The other warning is that hydrogen atoms were found; these usually cannot be refined unconstrained with x-ray data. The warning recommends replacing them with stereochemically determined ones (this is illustrated in the 3rd Exercise of this tutorial) and the last line of the Warning tells you what to do if they are to be refined. Here we will only refine twin laws & scale factors so this is unnecessary. Press OK; you get the opportunity to change the Phase name (I know it is very long but that doesn’t matter here). Press OK; the General tab for this phase is shown.
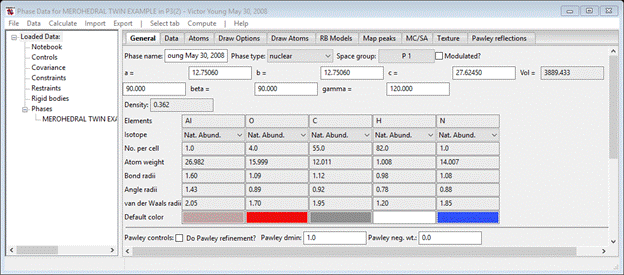
Change the Space group to P 32; the window will be redrawn after a popup showing the operations. You can see the structure by selecting Draw Atoms.
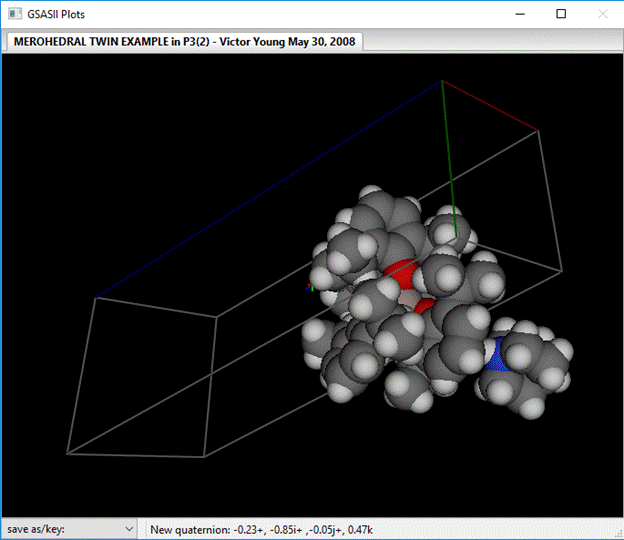
The large anion containing the Al atom is to the left and the smaller cation to the right.
Next do Import/Structure Factor/from Shelx HKLF 4 F2 file and select 2b.hkl from the file dialog box; a popup will appear showing a few lines.

This is the right file; a Shelx HKLF 4 file consists of records with just h,k,l, F2 and s(F2). Press Yes and OK on the next one (we’ll keep the name as it is). Choose the phase with the long name on the next popup and press OK; select the Structure factors tab, a 2D display of the hk0 zone is plotted.
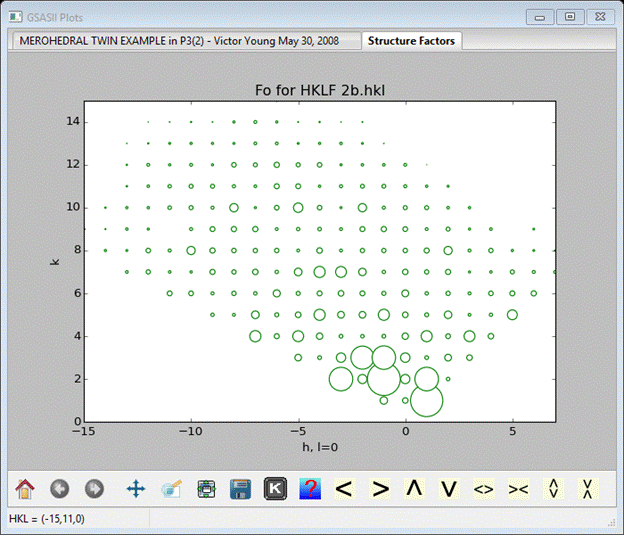
The size of the green circles are proportional to Fo (after a structure factor calculation blue rings will appear for Fc).
You now have loaded all that is needed for the second example. The structure had previously been solved and refined with Shelx so you need not trouble about those steps. You can try to solve the structure in GSAS-II via charge flipping but this is not part of this exercise (it isn’t easy because of the twinning); we will only explore the twin analysis features of GSAS-II.
Step 2. Refinement
of twinned structure
Before beginning the refinement, it usually the practice to eliminate the poorly measured reflections where F2obs < 2s(F2). Select the Controls entry from the GSAS-II data tree.

Change Min obs/sig to 2.0. Next, select the Phases/MEROHEDRAL… entry in the GSAS-II data tree and choose the Data tab.
Notice that the Scale factor is selected for refinement by default. If you refine it, you will get an Rw~ 25% which is terrible. If you try refining atom positions & thermal parameters you could possibly lower it to ~15% (still terrible) and the resulting structure would not be very satisfactory. However, we have suspected from the equivalence of -3 and -3m1 Laue data symmetries that there is twinning so don’t bother trying to make it better by refining the structure. To determine the possible twin law we refer to Table 1.3.4.2 of the International Tables for Crystallography Vol C. and see that indeed Laue -3m1 as P3221 can be simulated by twinned Laue -3 crystals in P32; the possible operator is shown in Table 1.3.4.1 (reproduced above) as either a mirror or a 2-fold. The detailed symbolism in the table (‘m.., ..2/.2.’) indicates (perhaps obscurely) the operator specifics. See P3m1 and P3221 space groups in International Tables for Crystallography, Vol A for the operations m as -y-xz or 2 as yx-z; we will use the 2-fold since the m inverts the structure which we are not sensitive to in this experiment.
Select Add Twin Law; the Data tab will be redrawn with the inversion for the second law. Replace this with the 2-fold (0 1 0, 1 0 0, 0 0 -1) and check the Refine box. It should look like.
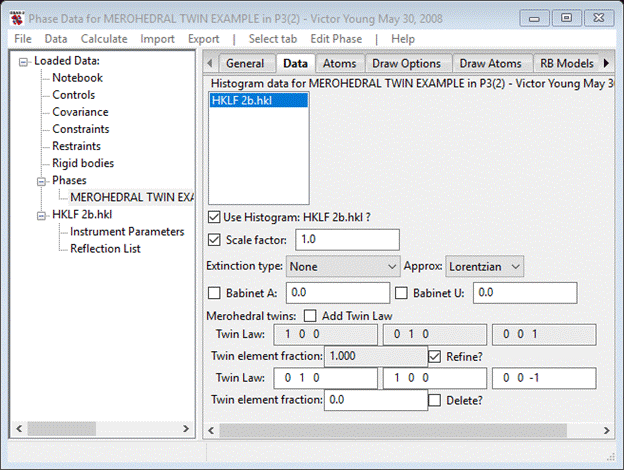
Now do Calculate/Refine; you’ll have to save the project. Pick a directory & file name - I used 2b. there will be an immediate improvement in the fit to Rw=5.05% and the twin fractions will be ~50/50. Clearly this crystal is Type 2 merohedrally twinned; we cannot determine if there is any Type 1 twinning because we lack sufficient resonant scattering to see it. This ends the second example; you can save the project file.
Example 3. A Type 1
& 2 merohedral twin – Cp*Cs(NC5H5)
This ecample is very similar to the previous one except that the Cs atom has significant resonant scattering for MoKa radiation allowing detection of Type 1 merohedry. The data set has apparent 6/m Laue symmetry but may be 6/mmm for the space groups P61/P65 or P6122/P6522. The combination of Type 1 and Type 2 twins requires 4 twin laws to be explored for this pentamethylcyclopentadienyl Cs complex. As this work was done with Shelx this result is in the form of a Shelx input file (3a.ins – thanks to V. Young for this example). If you have not done so already, start GSAS-II and start with a fresh project.
Step 1. Setup for
refinement
To begin we need to import the phase information from the Shelx ins file. From the main GSAS-II data tree menu, do Import/Phase/from Shelx ins, res file (the ins file is input to Shelx and a res file is the output result after a Shelx run; either can be used here). Select 3a.ins from the file selection window; a popup window will ask if this file is the one you want.
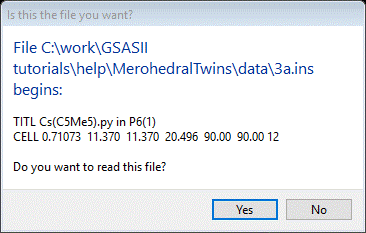
It is; press the Yes button. You will then see the SHELX Read Warning; you will have to change the space group to P61 in a bit. Press OK and leave the name alone (press OK again). Change the Space group to P 61; the General tab will be redrawn after showing you the operators.

Next do Import/Structure Factor/from CIF file and select 3a.fcf from the file dialog. Press Yes in the next popup and OK on the name change popup. Select the file name in the Add to phases popup and press OK; the hk0 zone of the structure factors will be displayed. Note that this plot shows both green (Fo) and blue (Fc) circles with a small solid red or green circle in the centers. These are for the +/- DF values. Fc is here because this fcf file was obtained after refinement via Shelx.
You now have loaded all that is needed for the third example. The structure had previously been solved and refined with Shelx so you need not trouble about those steps. You can try to solve the structure in GSAS-II via charge flipping but this is not part of this exercise (it isn’t easy because of the twinning; charge flipping will readily give the Cs position & most of the rest. It can be completed via difference Fourier techniques); we will only explore the twin analysis features of GSAS-II.
Step 2. Refinement
of twinned structure
Before beginning the refinement, it usually the practice to eliminate the poorly measured reflections where F2obs < 2s(F2). Select the Controls entry from the GSAS-II data tree.
Change Min obs/sig to 2.0. Next, select the Phases/Cs… entry in the GSAS-II data tree and choose the Data tab.

Notice that the Scale
factor is selected for refinement by default. If you refine it, you
will get an Rw~ 17% which is terrible. It
doesn’t change much if you add the Flack parameter (~0.36). If you try refining
atom positions & thermal parameters you could possibly lower it but the
resulting structure would not be very satisfactory. However, we have suspected
from the equivalence of 6/m and 6/mmm Laue data symmetries that there is
twinning so don’t bother trying to make it better by refining the structure. To
determine the possible twin laws we again refer to Table 1.3.4.2 of the
International Tables for Crystallography Vol C. and see that indeed Laue 6/mmm
as P6122 can be simulated by twinned Laue 6/m crystals in P61;
the possible operator is shown in Table 1.3.4.1 (reproduced above) as either a
mirror or a 2-fold. The detailed symbolism in the table (‘m.., ..2/.2.’)
indicates (perhaps obscurely) the operator specifics; they are m as ![]() or
2 as
or
2 as ![]() ; we will use the 2-fold, m and
inversion operators to fully test for presence of the 4 possible twin laws.
Press the Add Twin Law button 3 times; each time the window will be
redrawn. When done you should see.
; we will use the 2-fold, m and
inversion operators to fully test for presence of the 4 possible twin laws.
Press the Add Twin Law button 3 times; each time the window will be
redrawn. When done you should see.
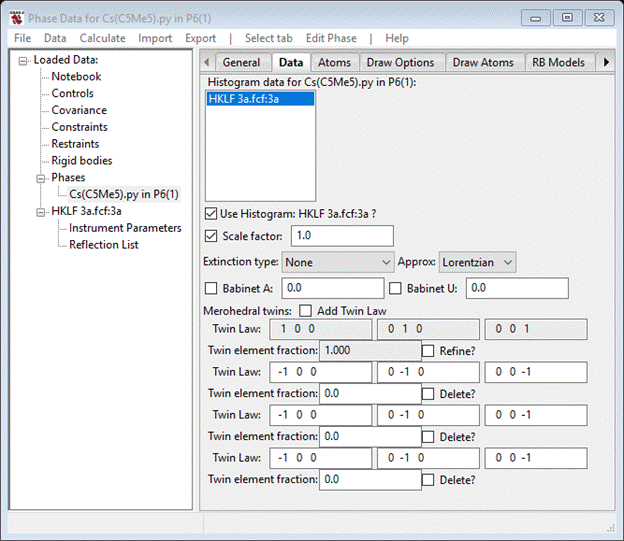
For the third and fourth matrix enter 0 1 0 1 0 0 0 0 -1 and 0 -1 0 -1 0 0 0 0 1 as the 2-fold and mirror operators (the 2nd matrix is the inversion by default). Select the Refine button and do Calculate/Refine from the main menu; again you’ll have to save the project file – I called it 3a. The Rw will immediately drop to ~2.26% (RF~2.05%). The four twin element fractions will show various values
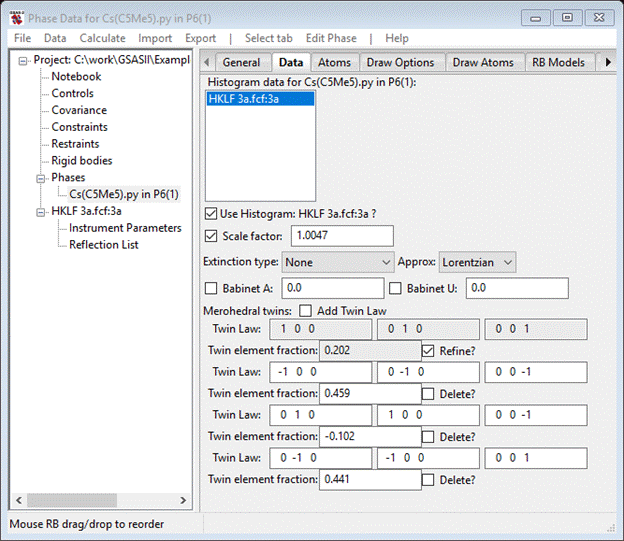
The negative one for the 3rd twin fraction is nonphysical and the 1st twin fraction is small. It is likely that the true space group is P65 and Type 2 merohedrally twinned (twin fractions 2 & 4). We can improve it by refining the atom positions and thermal parameters, but first we need to replace the hydrogen atoms as suggested by the Shelx warning. Select the Atoms tab for the phase.
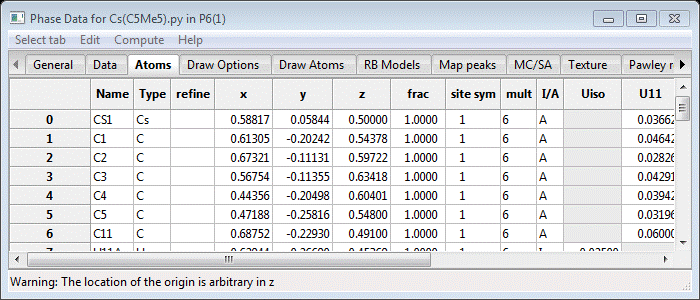
If you scroll down the page, you’ll see H-atoms with default Uiso (=0.025) values. To replace them, the old ones need to be deleted; double click the Type column heading and select H and press OK. The H-atoms will be highlighted (and plotted in green). Do Edit Atoms/On selected atoms…/Delete atom from the menu; the H-atoms will be deleted and the structure will be redrawn. Next double click the Type column heading and select C; all the C-atoms will be selected (and shown in green on the plot). Do Edit Atoms/On selected atoms…/Calc H atoms; a Distance Angle Controls will appear. Occasionally these values may need to be changed but not this time; press Ok. The next popup is for the H atoms add controls.

It specifies the expected geometry around each C-atom for H-atom placement. As expected we want 3 H-atoms on the terminal CH3 groups on the Cp* ring and one for each C-atom for the NC5H5 molecule. Press Ok; you will see the H-atoms added in turn to the structure and they will be inserted immediately after their respective C-atom. Their positions and thermal parameters are tied to the C-atoms so that after refinement of the structure they can be shifted to the stereochemically best positions. For the refinement, double click the Refine column heading and select X and U from the popup; press OK. The Atom table will be redrawn with XU in every entry under Refine. Now do Calculate/Refine (2-3 times until convergence)from the main GSASII data tree window; the Rw will drop to ~1.80%. If you look at the Data tab the Twin element fractions are much clearer.

The identity Twin Law has virtually vanished along with the 2-fold one leaving just the inverse and mirror Twin Laws.
This means the structure is the inverse in space group P65 but still Type 2 merohedrally twinned.
To test this, we need to both change the space group and invert the structure. First select the General tab and change the space group to P 65; you’ll see the Operators popup (press Ok) and the General tab will be redrawn. Next select the Atoms tab; do Edit Atoms/Update H atoms to put them in their best positions. Next select all the atoms by double clicking the empty box in upper left corner of the table; all atoms will be highlighted & plotted green. Do Edit Atoms/On selected atoms…/Transform atoms; a popup will appear.
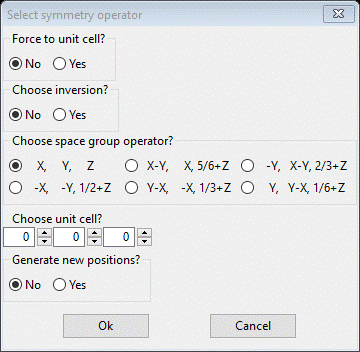
Check the Choose inversion box and (important) set Choose unit cell to 1 1 1; this will put the inverted structure back inside the unit cell. Press Ok; the table and plot will both be redrawn. Now do Calculate/Refine 2-3 times; the Rw will drop back to where it was before and the Data tab will show new Twin fractions.

Now the identity Twin Law is ~2/3 and the 2-fold Twin Law is the other ~1/3 while the other two are ~zero. You can delete them and repeat the refinement; Rw ~1.7%. This completed the merohedral twin exercises. You can save this project if desired.